

Introduction
Materials and Methods
Plant materials
DNA extraction and whole genome sequencing data
cpGenome sequence assembly
Sequence alignment and phylogenetic analysis
Sequence identity analysis
Variant calling and haplotype inference for cpGenome heteroplasmy
Results
Complete cpGenomes of five Korean landraces
Phylogenetic analysis based on whole cpGenome sequences
cpGenome identity relative to ‘Zea perennis’
Quantification of cpGenome heteroplasmy across Korean landraces
Discussion
Introduction
Chloroplast genomes (cpGenomes) offer a valuable resource for understanding plant evolution, domestication, and adaptation. Due to their relatively small size, conserved quadripartite structure, and predominantly maternal inheritance, cpGenomes have long been used to resolve phylogenetic relationships, investigate population structure, and track crop dispersal (Palmer, 1985; Birky, 2001; Gitzendanner et al., 2018). In addition to their core roles in photosynthesis and biosynthesis, variation in cpGenomes can reflect lineage-specific evolutionary histories and may harbor traits of agronomic relevance (Daniell et al., 2016).
Maize (Zea mays L.) is a major cereal crop of global economic importance and a model system for plant genetics and genomics. A significant component of its genetic diversity is maintained in traditional landraces, which have been cultivated in diverse agroecological regions over centuries. These landraces retain allelic and structural variations that are often absent from modern inbred lines, making them valuable genetic resources for crop improvement and for understanding domestication, adaptation, and historical dispersal processes (Matsuoka et al., 2002; Camus-Kulandaivelu et al., 2006; Hufford et al., 2012). Despite their relevance, cpGenomes-level diversity among maize landraces remains underrepresented in current genomic datasets. While the nuclear genome of maize has been extensively characterized, the cpGenome has received comparatively less attention, especially in the context of landraces. In recent years, the number of publicly available maize cpGenomes has rapidly increased (Wang et al., 2024), enabling comprehensive comparative studies across modern inbred lines and landraces from different regions (López et al., 2021). However, the extent to which chloroplast genome diversity contributes to the adaptation and diversification of landraces remains poorly understood. Despite this growing dataset, Korean maize landraces remain largely unexplored at the cpGenome level, and their maternal origins and evolutionary relationships have not been characterized.
To address this gap, we assembled the complete cpGenomes of five uncharacterized Korean maize landraces and compared them with publicly available cpGenome sequences. We further explored their phylogenetic relationships and investigated heteroplasmic variation across the cpGenomes using variant allele frequency (VAF) profiles. The findings offer a basis for understanding the potential maternal background and plastome-level variation among Korean maize landraces. By characterizing cpGenome diversity and heteroplasmy patterns, this work provides foundational insights for future studies of maize cytoplasmic diversity and cpGenome evolution.
Materials and Methods
Plant materials
Five Korean maize landraces -DS302, DS553, DS1392, DS1563, and DS1942 (Table 1)- were obtained from the National Agrobiodiversity Center (https://genebank.rda.go.kr). The landraces all have flint kernels and were collected in 1980s, before commercial cultivars became widespread. Those plants were cultivated in the experimental field of the Chungnam National University (Daejeon, Korea; 36.36°N, 127.35°E).
DNA extraction and whole genome sequencing data
Genomic DNA was extracted from fresh leaves of five maize landraces (Table 1) using the cetyltrimethylammonium bromide (CTAB) method (Doyle and Doyle, 1987). DNA quality and concentration were assessed using a Nanodrop spectrophotometer (Thermo Fisher Scientific) and integrity was confirmed by agarose gel electrophoresis. A paired-end genomic library with an insert size of 151 bp was constructed and sequenced on the Illumina NovaSeq 6000 platform at JS Link (Seoul, Korea).
cpGenome sequence assembly
Raw whole-genome sequencing reads from five maize landraces were quality-trimmed using Trimmomatic v0.36 (Bolger et al., 2014). High-quality reads were then aligned to the reference cpGenome sequence of B73 (KF241981.1) using BWA (Li and Durbin, 2009). Alignments that were unmapped or not primary were filtered out with SAMtools view (Danecek et al., 2021) using the -F 260 to exclude unmapped and secondary alignments. Filtered reads were subsequently used for cpGenome assembly, which was performed with GetOrganelle v1.7.4.1 (Jin et al., 2020), employing a range of k-mer values (-k 21, 29, 39, 45, 59, 65, 79, 85, 99, 105, 119) and 20 iterative assembly rounds (-R 20) to optimize circular genome reconstruction. A circular genome map was created using OrganellarGenomeDRAW v1.3 (Greiner et al., 2019).
Sequence alignment and phylogenetic analysis
To enable high-quality comparative analysis, cpGenome sequences from ten maize accessions available in the NCBI database (Table 1) were aligned alongside those of the Korean landraces using MAFFT v7.511 (Yamada et al., 2016). Phylogenetic reconstruction was performed with IQ-TREE 2 (Nguyen et al., 2015) under default settings, using ‘Zea perennis’ as an outgroup.
Table 1.
Maize samples used in this study.
Sequence identity analysis
Total of five cpGenomes (DS302, DS553, DS1392, B73, and ‘Zea perennis’) were aligned using MAFFT v7.511, followed by a sliding window-based sequence identity analysis. Pairwise identity between ‘Zea perennis’ and each aligned sequence was calculated with a window length of 500 bp and a step size of 100 bp using a custom R script. The resulting identity values, ranging from 95 to 100%, were visualized with the ggplot2 (Wickham, 2011) package in R.
Variant calling and haplotype inference for cpGenome heteroplasmy
BAM files previously aligned to the B73 cpGenome were directly used for variant calling. Variants were identified using GATK HaplotypeCaller (McKenna et al., 2010) with the following parameters: --min-base-quality-score 30 and --output-mode EMIT_ALL_ACTIVE_SITES. Reference confidence output was set to GVCF, and soft-clipped bases were excluded. During alignment and variant calling, reads originating from inverted repeat (IR) regions may have been mapped ambiguously to multiple positions and subsequently excluded, resulting in a lack of called variants in those regions. Variant calls were filtered with GATK VariantFiltration with thresholds of QUAL < 30.0 and MQRankSum < −12.5. Biallelic single nucleotide polymorphisms (SNPs) that passed the filters were extracted using bcftools (Danecek et al., 2021), and allelic depths were exported in tabular format. VAF value was calculated as the alternative allele depth divided by total depth. VAF values were summarized using a sliding window approach (window length = 1,000 bp; step size = 200 bp) in R, and the average VAF per window was visualized using ggplot2.
Results
Complete cpGenomes of five Korean landraces
The complete cpGenomes of five Korean maize landraces were successfully assembled through reference-guided assembly using high-quality Illumina reads, which provided an average of approximately 600,000 reads covering 100% of the B73 reference cpGenome with a mean depth of 7,000×. All Korean landrace cpGenomes exhibited the typical quadripartite structure (Fig. 1), consisting of a large single-copy (LSC) region, a small single-copy (SSC) region, and a pair of IRs, and had an average size of approximately 140 kb as previously reported (Maier et al., 1995). When each accession was individually analyzed, the total genome size was highly conserved among the landraces, with DS1392 having a length of 140,450 bp and the others measuring 140,453 bp (Table 2). The overall GC content was also similar across accessions: 38.437% in DS302, DS1392, and DS1942, and 38.436% in DS553 and DS1563.
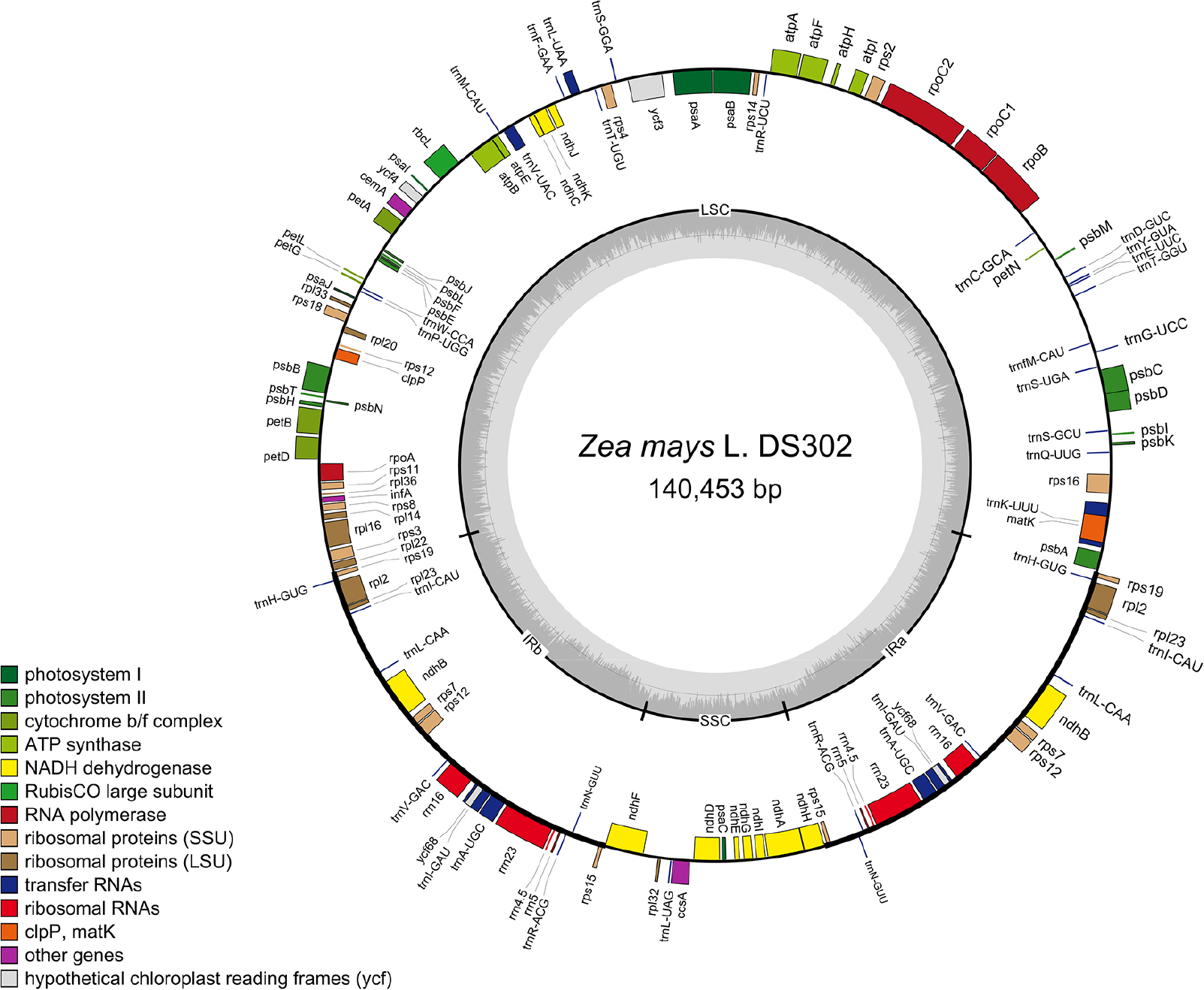
Fig. 1.
Circular chloroplast genome (cpGenome) map of the DS302. Genes transcribed in the clockwise direction are shown on the inner side of the circle, and those transcribed counterclockwise on the outer side. Genes are color-coded by functional categories as designated. The inner gray plot indicates GC content. LSC, large single-copy; IRb, inverted repeat b; SSC, small single-copy; IRa, inverted repeat a.
Table 2.
Statistics of the assembled chloroplast genomes (cpGenomes).
Phylogenetic analysis based on whole cpGenome sequences
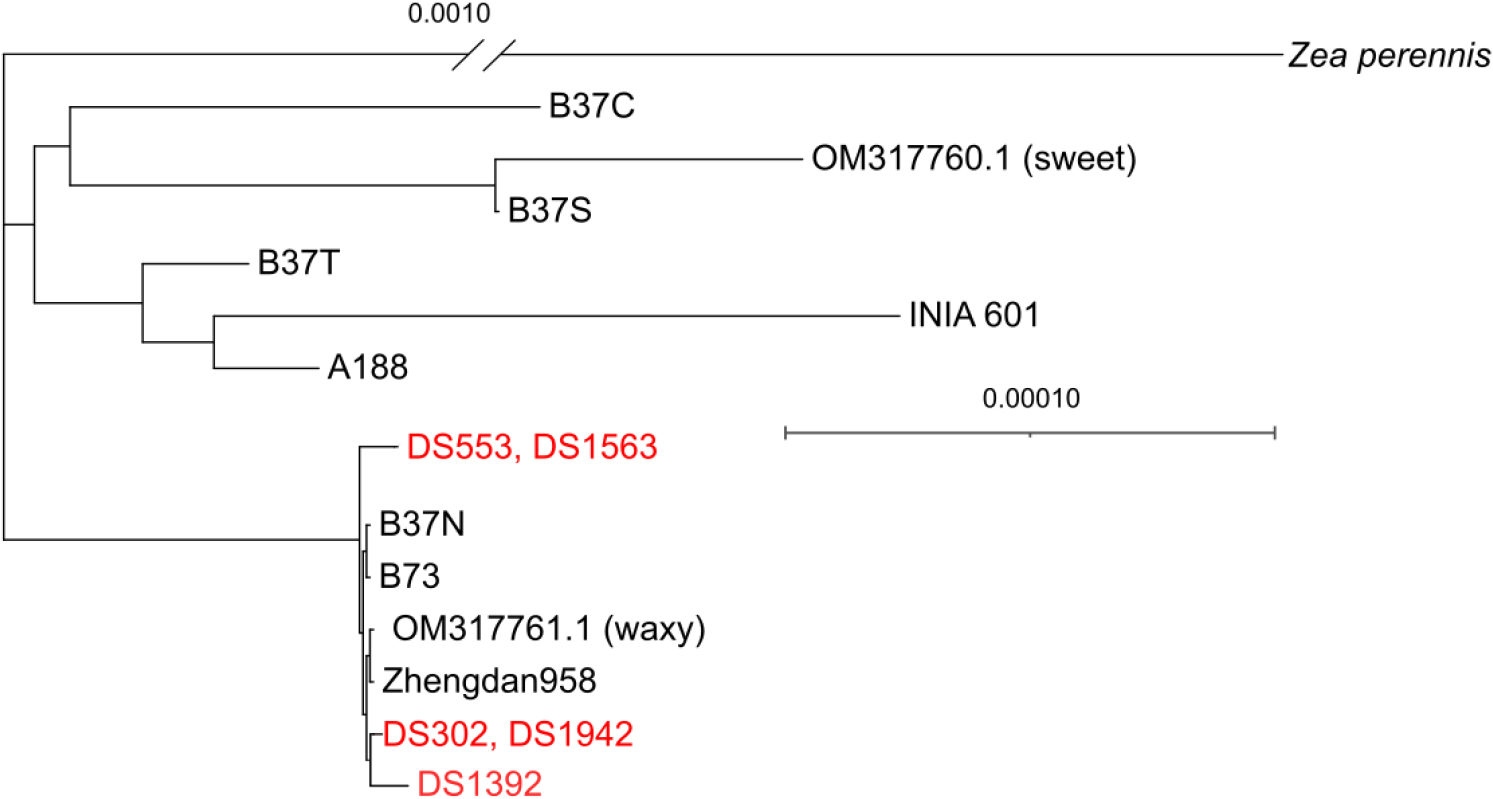
To clarify the phylogenetic position of the Korean landraces, complete cpGenome sequences of ten Z. mays accessions and one ‘Zea perennis’ (used as an outgroup) available from NCBI database were included in the phylogenetic analysis (Table 1). Among the Korean landraces, four accessions except DS1392 shared identical cpGenome sequences in pairs: DS553 with DS1563, and DS302 with DS1942 (Fig. 2). It was observed that DS302 and DS1942 exhibited shorter branch lengths compared to the other Korean landraces, suggesting minimal sequence divergence within this subgroup. Across the phylogenetic tree, ‘Zea perennis’ and Z. mays were clearly separated, and INIA 601, which is closely related to Z. mays ssp. parviglumis, was positioned closer to ‘Zea perennis’ than to the other Z. mays accessions (Montenegro et al., 2022; Yang et al., 2023). In contrast, all Korean landraces were grouped into a single clade and clustered with modern inbred lines such as Zhengdan958 and B73, forming the largest clade observed in the analysis. This result suggests that the maize introduced to Korea was already in an advanced state of domestication (Yi et al., 2019). Furthermore, the Korean landraces formed a closer subclade with the Chinese inbred line Zhengdan958 than with B73.
cpGenome identity relative to ‘Zea perennis’
Pairwise sequence identity was calculated using ‘Zea perennis’ as a reference, with a window length of 500 bp and a step size of 100 bp, based on three Korean landraces accessions with distinct cpGenome sequences and the inbred line B73 (Fig. 3). To enable clearer visual comparison of the differences between B73 and the Korean landraces, the y-axis range for identity was limited to 95 - 100%. The LSC region exhibited the greatest divergence between ‘Zea perennis’ and Z. mays accessions. It was observed that the overall divergence patterns from ‘Zea perennis’ were similar among all Z. mays accessions analyzed. When comparing the Korean landraces to B73, the most pronounced differences were observed near the 16 kb and 62 kb positions, where identity decreased to approximately 95%, representing the greatest divergence from B73. Comparison of aligned sequences revealed that these regions contained indels associated with poly-T repeat sequences.
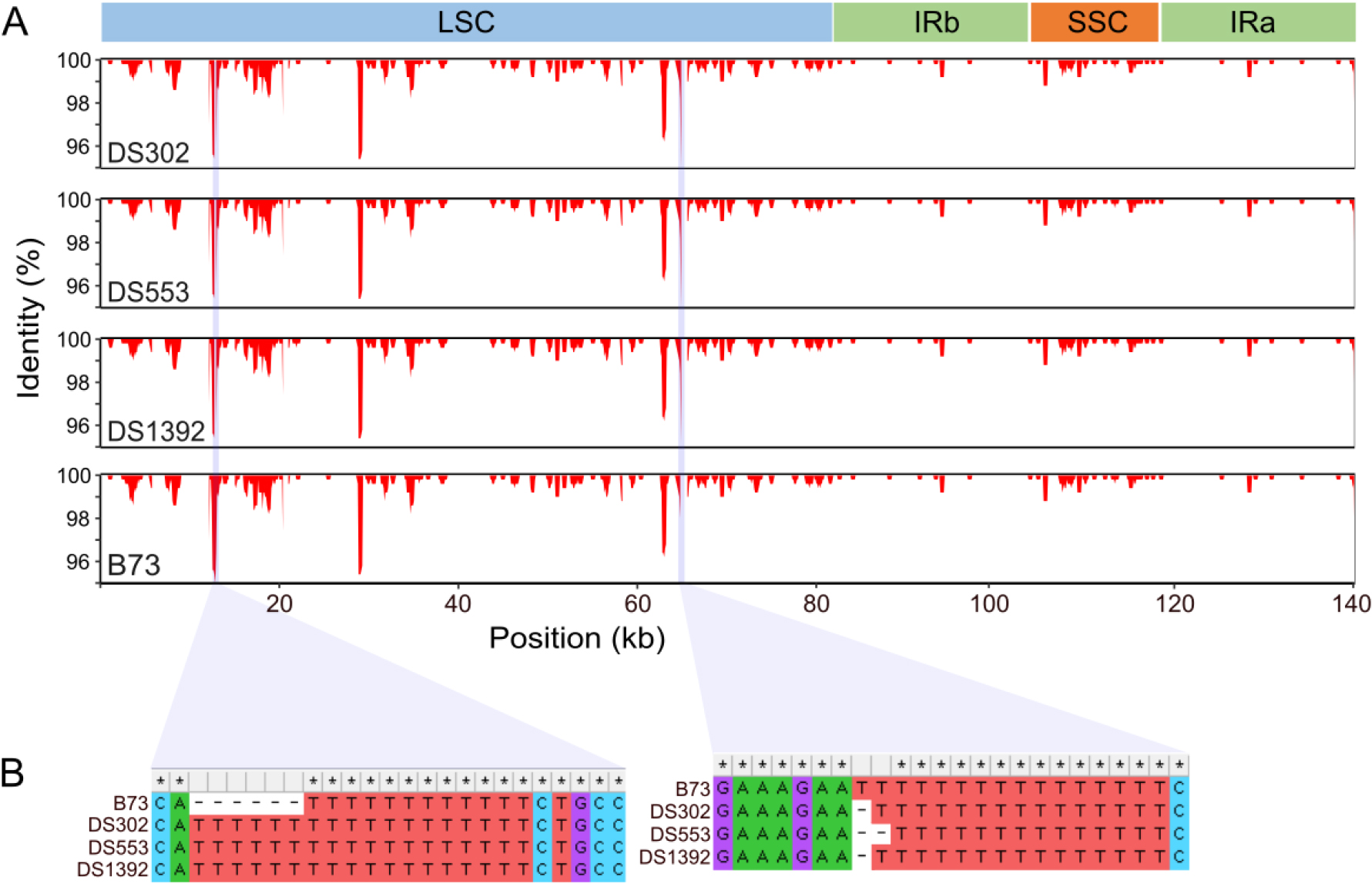
Fig. 3.
Identity plot of four Z. mays accessions relative to ‘Zea perennis’, used as the reference. (A) Identity values were calculated using a sliding window approach (window length = 500 bp; step size = 100 bp), and are shown in the range of 95 to 100%. Purple-shaded regions indicate the most divergent sequences (B). Aligned sequences corresponding to these regions, generated using MEGA11. LSC, large single-copy; IRb, inverted repeat b; SSC, small single-copy; IRa, inverted repeat a.
Quantification of cpGenome heteroplasmy across Korean landraces
Organelle genomes are known to exhibit heteroplasmy, characterized by the presence of multiple plastid types, often resulting from biparental inheritance (Wolfe and Randle, 2004; Ramsey and Mandel, 2019; Sawicki et al., 2023). To quantify heteroplasmy levels in Korean maize landraces, allele depth imbalance across chloroplast loci was assessed with a window length of 1,000 bp and a step size of 200 bp (Fig. 4). Among the five accessions, DS1942 exhibited the highest level of chloroplast heteroplasmy, with 375 heteroplasmic positions identified. The other accessions showed varying numbers of differing positions: DS302 had 45, DS553 had 76, DS1392 had 216, DS1563 had 90, and B73 had 359 (Table 3). The average VAF among all Korean landrace accessions ranged from 0.017 to 0.032, with DS302 exhibiting the highest mean VAF. B73 showed a VAF of 0.02, ranking as the third lowest among all tested accessions. The 57 - 58 kb position in the LSC region exhibited elevated heteroplasmy across all accessions, while the 104 kb position in the SSC region showed increased VAF in all accessions except B73. The 57 - 58 kb region corresponded to the rbcL gene, while the 104 kb region was located within a non-coding intergenic region. Although DS553 and DS1563, which share identical cpGenome sequences, showed similar heteroplasmy profiles, DS302 and DS1942 diverged significantly in their heteroplasmic patterns. In particular, DS1942 displayed widespread heteroplasmy throughout the cpGenome, indicating a relatively high level of intra-individual sequence heterogeneity.
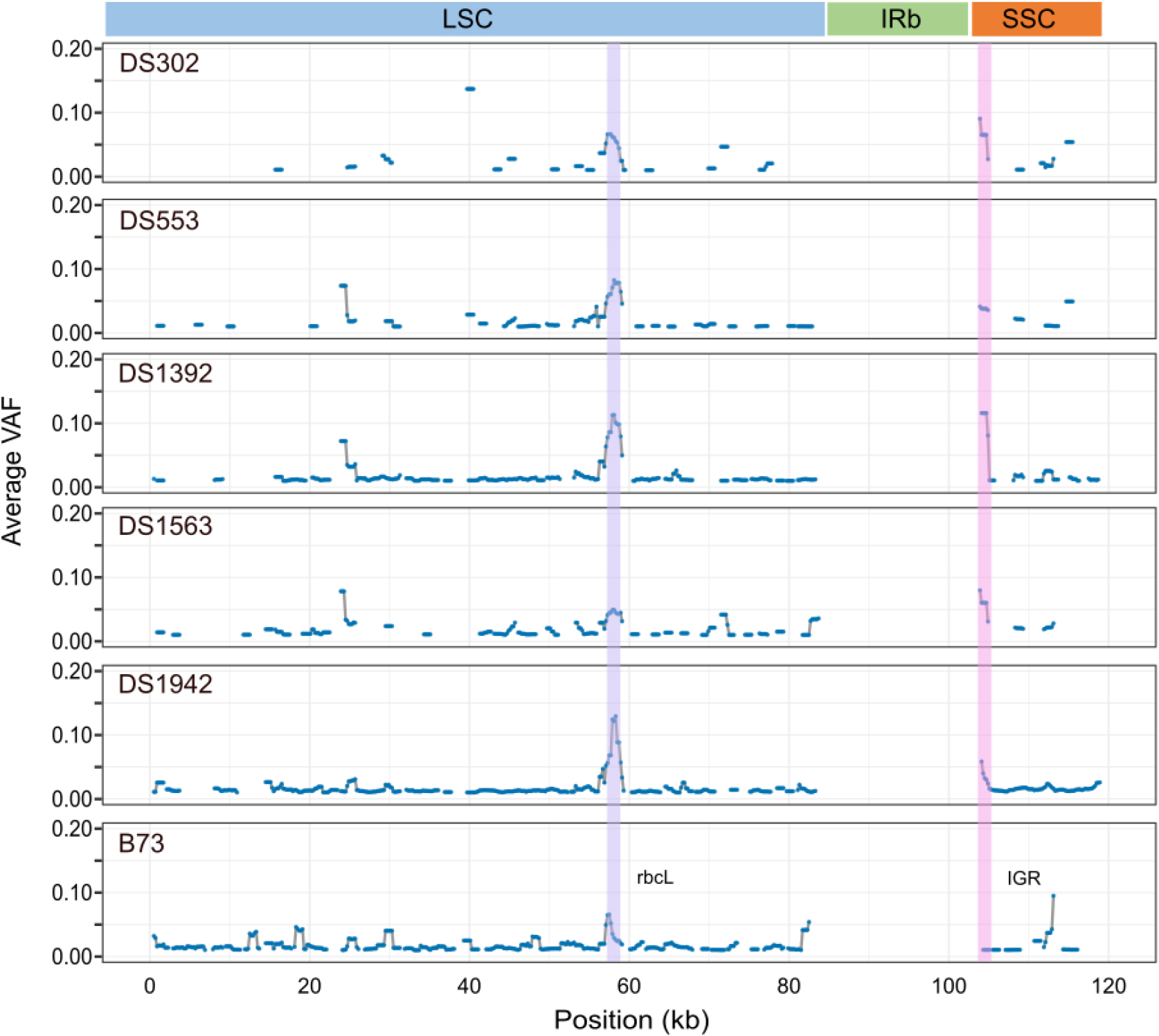
Fig. 4.
Visualization of heteroplasmy levels across the chloroplast genome (cpGenome) based on variant allele frequency (VAF). Heteroplasmic sites were identified using GATK HaplotypeCaller and filtered with VariantFiltration (QUAL < 30.0 and MQRankSum < -12.5), followed by an additional depth filter (depth > 50). Only sites with VAF > 1% were included in the analysis. VAF values were summarized using a sliding window approach (window length = 1,000 bp; step size = 200 bp). LSC, large single-copy; IRb, inverted repeat b; SSC, small single-copy; IGR, intergenic region.
Discussion
In this study, we assembled and characterized the complete cpGenomes of five Korean maize landraces (Table 1) to explore their structural and sequence-level variations. The overall quadripartite structure and genome size of the landrace cpGenomes were largely conserved, consistent with previous reports in maize and other grasses (Hiratsuka et al., 1989; Maier et al., 1995). Phylogenetic analysis using complete cpGenome sequences, combined with publicly available data (Table 1). Our findings reaffirm the deep evolutionary divergence between Z. perennis and Z. mays, consistent with previous nuclear-genome-based studies, underscoring their clear phylogenetic distinction (Fig. 2; Chen et al., 2022). Moreover, publicly available cpGenomes included in our analysis exhibited phylogenetic relationships identical to those observed in previous studies (Bosacchi et al., 2015). The concordance between our findings and previous reports provides strong support for the accuracy and reliability of our cpGenome assemblies (Bosacchi et al., 2015; Chen et al., 2022). Phylogenetic analysis further revealed that the Korean landraces grouped with modern inbred lines such as B73 and Zhengdan958 (Fig. 2). This pattern suggests that the maternal lineage of the Korean landraces was likely derived from already domesticated germplasm, implying their introduction to Korea occurred after domestication events (Yi et al., 2019).
Although the cpGenome sequences of these landraces were nearly identical across accessions (Fig. 3), analysis of heteroplasmy revealed considerable differences among them (Fig. 4). Heteroplasmy, the presence of more than one type of organellar genome within an individual, can arise through various mechanisms including biparental inheritance, replication-associated mutations, or bottlenecks during organelle transmissions (Wolfe and Randle, 2004; Ramsey and Mandel, 2019; Sawicki et al., 2023). Although the overall VAF values were low, all five Korean landraces displayed detectable signatures of chloroplast heteroplasmy (Fig. 4). In landrace populations that are not yet genetically fixed, heteroplasmy has been reported to increase following biparental transmission and persist over several generations before eventual sorting or loss (Wolfe and Randle, 2004; Greiner et al., 2015; Li and Cullis, 2023; Sawicki et al., 2023). Consistent with this, we observed higher VAF mean value in DS302, DS553, DS1563 (Table 3) compared to the NAM inbred B73 (Fig. 4) which could be indicative of incomplete sorting of ancestral cpGenome variants.
These findings suggest that the Korean landraces are more closely related to Chinese germplasm. Minor allele depth analysis also indicated that a subset of landraces may possess higher levels of chloroplast heteroplasmy than inbred lines, potentially reflecting residual signals from unidentified donor lineages. However, given the limited number of accessions, the lack of pedigree information, and the limited availability of whole-genome sequencing data with comparable depth levels, further validation using a broader panel of landraces and expanded sampling is required to confirm these patterns and clarify their evolutionary implications.
